Bioinformatics Toolbox
Lire, analyser et visualiser des données génomiques et protéomiques
Vous avez des questions ? Contacter un commercial.
Vous avez des questions ? Contacter un commercial.
Bioinformatics Toolbox propose des algorithmes et des applications pour le séquençage de nouvelle génération (NGS), l'analyse des biopuces, la spectrométrie de masse et l'ontologie génétique. Avec les fonctions de la toolbox, vous pouvez lire des données génomiques et protéomiques à partir de formats de fichier standard tels que SAM, FASTA, CEL et CDF, et à partir de bases de données en ligne, comme NCBI Gene Expression Omnibus et GenBank®. Vous pouvez parcourir et visualiser ces données avec des navigateurs de séquences, des cartes thermiques spatiales et des clustergrams. La toolbox propose également des techniques statistiques pour la détection de pics, la saisie de valeurs pour les données manquantes et la sélection de caractéristiques.
Vous pouvez combiner les fonctions de la toolbox pour supporter les processus bioinformatiques courants. Vous pouvez utiliser des données ChIP-Seq pour identifier les facteurs de transcription, analyser des données de séquences d'ARN pour identifier des gènes exprimés de manière différentielle, identifier des variantes de nombre de copies et des SNP dans des données de biopuces, et classifier des profils protéiques à partir de données de spectrométrie de masse.
En savoir plus sur la biologie computationnelle.
Bioinformatics Toolbox propose des algorithmes et des techniques de visualisation pour l'analyse de séquençage de nouvelle génération. La toolbox vous permet d'analyser des génomes dans leur intégralité, tout en effectuant des calculs à un niveau de résolution de paires de base. Vous pouvez utiliser le navigateur NGS pour visualiser et étudier des alignements à lecture courte à partir de lectures courtes en paire ou individuelles. Vous pouvez également créer des routines d'analyse personnalisées, comme indiqué dans les exemples suivants.
Les jeux de données utilisés dans l'analyse de séquençage de nouvelle génération sont généralement trop volumineux pour être stockés dans la mémoire physique. Bioinformatics Toolbox fournit des conteneurs de données dédiés qui vous permettent d'analyser des génomes dans leur intégralité.
L'objet BioIndexedFile vous permet d'accéder aux contenus de fichiers texte contenant des entrées de taille non uniforme telles que des séquences, des annotations et des références croisées au jeu de données. Vous pouvez générer ces objets à partir de tableaux, de fichiers non hiérarchiques ou de formats spécifiques à une application (SAM, FASTA, FASTQ, etc.).
La classe BioMap enregistre des informations de séquences à lecture courte, notamment des en-têtes de séquences, des lectures de séquences, des scores de qualité ainsi que des données d'alignement et de cartographie à une séquence de référence simple. Vous pouvez utiliser des méthodes et des propriétés d'objet pour parcourir, accéder, filtrer et manipuler les données contenues dans un objet BioMap.
Vous pouvez utiliser différentes méthodes pour normaliser des données de biopuces, comme la régression locale, la moyenne générale, l'écart absolu médian (MAD) et la normalisation quantile. Vous pouvez appliquer ces méthodes à l'ensemble de la biopuce, ou à des régions ou des blocs spécifiques. Les fonctions de filtrage et d'imputation vous permettent de nettoyer des données brutes avant d'exécuter les routines d'analyse et de visualisation.
Bioinformatics Toolbox vous permet d'effectuer des ajustements en arrière-plan et de calculer des valeurs d'expression de gène (ensemble de sondes) à partir des données de sonde de biopuces Affymetrix®, à l'aide des procédures Robust Multi-Array Average (RMA) et GC Robust Multi-Array Average (GCRMA). Vous pouvez appliquer une segmentation binaire circulaire aux données CGH de puces et estimer le taux d'erreur de plusieurs hypothèses testant les données d'expression génétique d'une expérience de biopuces. Vous pouvez également effectuer une normalisation de jeux invariants au rang, sur les intensités des sondes pour plusieurs fichiers CEL Affymetrix, ou sur les valeurs d'expression génétique de deux conditions expérimentales distinctes.
Les routines spécialisées pour la visualisation de données de biopuces comprennent des tracés en volcan, des tracés en boîte, des tracés loglog, des tracés I-R et des cartes thermiques spatiales de la biopuce. Vous pouvez également visualiser des idéogrammes avec des modèles de bandes G.
En utilisant des routines de Statistics and Machine Learning Toolbox, vous pouvez classifier vos résultats, effectuer un partitionnement k-means ou hiérarchique, et représenter vos données de biopuces dans des visualisations statistiques, telles que des clustergrams 2D avec un ordonnancement optimal des feuilles, des cartes thermiques, des tracés des composants principaux et des arbres de classification.

Tracé en volcan de données de biopuces montrant la signification opposée au rapport d'expression génétique.
Bioinformatics Toolbox propose un ensemble de fonctions dédiées à l'analyse de données de spectrométrie de masse. Ces fonctions permettent le prétraitement, la classification et l'identification de marqueurs à partir de données SELDI, MALDI, LC/MS et GC/MS. Les fonctions de prétraitement comprennent la correction, le lissage, le calibrage et le rééchantillonnage de référence. Vous pouvez aligner des données spectrales brutes en utilisant l'axe M/Z et effectuer un alignement du temps de rétention sur des données LC/MS et GC/MS. Vous pouvez tracer plusieurs spectres simultanément.
Vous pouvez lisser, aligner et normaliser des spectres, puis utiliser les outils de classification et d'apprentissage statistique pour créer des classificateurs et identifier des biomarqueurs potentiels.

Analyse protéomique et métabolomique différentielle sans étiquette avec Bioinformatics Toolbox.
Bioinformatics Toolbox vous permet d'appliquer la théorie des graphes à des matrices creuses. Vous pouvez créer, visualiser et manipuler des graphes tels que des cartes interactives, des tracés hiérarchiques et des chemins. Vous pouvez identifier et visualiser les chemins les plus courts dans des graphes, tester des cycles dans des graphes orientés et déterminer l'isomorphisme entre deux graphes.
Bioinformatics Toolbox offre des fonctions qui reposent sur les algorithmes de classification et d'apprentissage statistique dans Statistics and Machine Learning Toolbox, notamment :

Apprentissage et visualisation statistiques.
Bioinformatics Toolbox vous permet d'accéder à la base de données d'ontologie génétique depuis MATLAB, d'analyser des fichiers annotés d'ontologie génétique et d'obtenir des sous-ensembles de l'ontologie tels que des ancêtres, des descendants ou des parents.
Bioinformatics Toolbox comprend des outils d'analyse et de visualisation de séquences pour les données de séquences génomiques et protéomiques. Vous pouvez réaliser diverses analyses, notamment des alignements de séquences multiples, ainsi que la construction, la visualisation interactive et la manipulation d'arbres phylogénétiques.
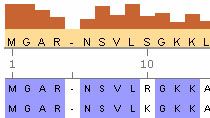
La toolbox propose des fonctions, des objets et des méthodes pour l'analyse de séquences, notamment l'alignement de séquences par paires, de profils de séquences et de séquences multiples. Ces fonctions incluent :
Voir les 3 images
La toolbox vous permet de manipuler et d'analyser vos séquences afin de mieux comprendre vos données. Vous pouvez :
La toolbox vous permet de visualiser des séquences et des alignements. Vous pouvez visualiser des cartes linéaires ou circulaires de séquences annotées avec des fonctionnalités GenBank. Vous pouvez visualiser des diagrammes de la structure secondaire d'une séquence d'ARN. Les afficheurs interactifs vous permettent de parcourir et de modifier des alignements de séquences multiples et par paires.
La toolbox vous permet de créer et de modifier des arbres phylogénétiques. Vous pouvez calculer des distances par paires entre des séquences de nucléotides ou d'acides aminés, qu'elles soient alignées ou non, en utilisant diverses mesures de similarité comme les méthodes de Jukes-Cantor, de la p-distance, du score d'alignement ou une méthode de distance définie par l'utilisateur. Les arbres phylogénétiques sont construits en utilisant une liaison hiérarchique avec une variété de techniques, y compris les méthodes du neighbor joining, de liaison simple et de liaison complète, et UPGMA (Unweighted Pair Group Method Average).
La toolbox permet de pondérer et de réenraciner des arbres, ainsi que de calculer des sous-arbres et la forme canonique des arbres. L'afficheur d'arbres phylogénétiques vous permet d'élaguer, de réorganiser et de renommer des branches, de parcourir des distances, ainsi que de lire ou d'écrire des fichiers au format Newick. Vous pouvez également utiliser les outils d'annotation dans MATLAB pour créer des arbres de qualité.
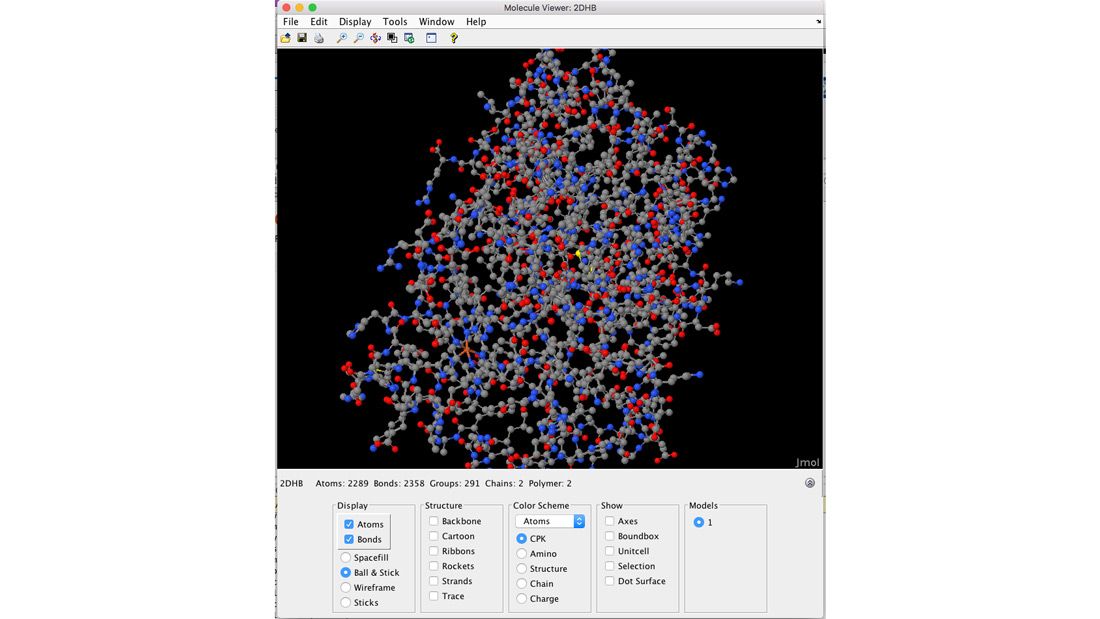
La toolbox offre des techniques d'analyse de séquences protéiques, notamment des routines pour le calcul de propriétés d'une séquence peptidique, telles que la composition atomique, le point isoélectrique et le poids moléculaire. Vous pouvez déterminer la composition en acides aminés des séquences protéiques, scinder une protéine avec une enzyme et créer des tracés de squelette et de Ramachandran de données PDB. Vous pouvez utiliser le Sequence Tool pour visualiser les propriétés d'une séquence d'acides aminés ou utiliser le Molecule Viewer pour afficher et manipuler des structures moléculaires en 3D.
Vous pouvez accéder aux formats de fichiers standard pour les données biologiques, les bases de données en ligne et les sites web. Bioinformatics Toolbox vous permet d’effectuer les opérations suivantes :
MATLAB propose des outils qui vous permettent de convertir votre programme d'analyse de données en une application logicielle personnalisée. Vous avez notamment accès à des outils de développement pour la création d'interfaces utilisateur, un environnement de développement visuel intégré et un profileur. Les produits de déploiement d'applications MATLAB vous permettent d'intégrer vos algorithmes MATLAB à des applications C, C++ et Java™, de déployer les algorithmes développés et les interfaces personnalisées sous forme d'applications autonomes, de convertir les algorithmes MATLAB en composants Microsoft® .NET ou COM accessibles depuis n'importe quelle application COM, ainsi que de créer des compléments Microsoft Excel®.
Vous pouvez intégrer MATLAB à des outils bioinformatiques couramment utilisés tels que BioPerl, des services Web basés sur SOAP et des plug-ins COM.

Partage d'algorithmes et déploiement d'applications.
Vous pouvez également sélectionner un site web dans la liste suivante :
Amériques
Europe
Asie-Pacifique