Using HMMs for Profile Analysis of a Protein Family
This example shows how HMM profiles are used to characterize protein families. Profile analysis is a key tool in bioinformatics. The common pairwise comparison methods are usually not sensitive and specific enough for analyzing distantly related sequences. In contrast, Hidden Markov Model (HMM) profiles provide a better alternative to relate a query sequence to a statistical description of a family of sequences. HMM profiles use a position-specific scoring system to capture information about the degree of conservation at various positions in the multiple alignment of these sequences. HMM profile analysis can be used for multiple sequence alignment, for database searching, to analyze sequence composition and pattern segmentation, and to predict protein structure and locate genes by predicting open reading frames.
Accessing PFAM Databases
Start this example with an already built HMM of a protein family. Retrieve the model for the well-known 7-fold transmembrane receptor from the Sanger Institute database. The PFAM key number is PF00002. Also retrieve the pre-aligned sequences used to train this model. More information about the PFAM database can be found at http://pfam.xfam.org/.
hmm_7tm = gethmmprof(2); seed_seqs = gethmmalignment(2,'type','seed');
For your convenience, previously downloaded sequences are included in a MAT-file. Note that data in public repositories is frequently curated and updated; therefore the results of this example might be slightly different when you use up-to-date datasets.
load('gpcrfam.mat','hmm_7tm','seed_seqs')
Models and alignments can also be stored and parsed in later directly from the files using the pfamhmmread, fastaread and multialignread functions.
Display the names and contents of the first three loaded sequences using the seqdisp command.
seqdisp(seed_seqs([1 2 3]),'row',70)
ans =
23×81 char array
'>VIPR2_HUMAN/123-371 '
' 1 YILVKAIYTL GYSVS.LMSL ATGSIILCLF .RKLHCTR.N YIHLNLFLSF ILRAISVLVK .DDVLYSSS.'
' 71 GTLHCPD... .......... .......... ....QPSSW. ..V.GCKLSL VFLQYCIMAN FFWLLVEGLY'
'141 LHTLLVA... ...MLPP.RR CFLAYLLIGW GLPTVCIGAW TAAR...... .........L YLED......'
'211 ......TGC. WDTN.DHSVP W....WVIRI PILISIIVNF VLFISIIRIL LQKLT..... .SPDVGGNDQ'
'281 SQY....... .......... .......... ....KRLAKS TLLLIPLFGV HYMV..FAVF PISI...S.S'
'351 KYQILFELCL GSF....QGL VV '
' '
'>VIPR_CARAU/100-348 '
' 1 FRSVKIGYTI GHSVS.LISL TTAIVILCMS .RKLHCTR.N YIHMHLFVSF ILKAIAVFVK .DAVLYDVIQ'
' 71 ESDNCS.... .......... .......... .....TASV. ....GCKAVI VFFQYCIMAS FFWLLVEGLY'
'141 LHALLAVS.. ...FFSE.RK YFWWYILIGW GGPTIFIMAW SFAK...... .........A YFND......'
'211 ......VGC. WDIIENSDLF W....WIIKT PILASILMNF ILFICIIRIL RQKIN..... .CPDIGRNES'
'281 NQY....... .......... .......... ....SRLAKS TLLLIPLFGI NFII..FAFI PENI...K.T'
'351 ELRLVFDLIL GSF....QGF VV '
' '
'>VIPR1_RAT/140-386 '
' 1 YNTVKTGYTI GYSLS.LASL LVAMAILSLF .RKLHCTR.N YIHMHLFMSF ILRATAVFIK .DMALFNSG.'
' 71 EIDHCS.... .......... .......... .....EASV. ....GCKAAV VFFQYCVMAN FFWLLVEGLY'
'141 LYTLLAVS.. ...FFSE.RK YFWGYILIGW GVPSVFITIW TVVR...... .........I YFED......'
'211 ......FGC. WDTI.INSSL W....WIIKA PILLSILVNF VLFICIIRIL VQKLR..... .PPDIGKNDS'
'281 SPY....... .......... .......... ....SRLAKS TLLLIPLFGI HYVM..FAFF PDNF...K.A'
'351 QVKMVFELVV GSF....QGF VV '
More information regarding how to store the profile HMM information in a MATLAB® structure is found in the help for hmmprofstruct.
Profile HMM Alignment
To test the profile HMM alignment tool you can re-align the sequences from the multiple alignment to the HMM model. First erase the periods in sequences used to format the downloaded aligned sequences. Doing this removes the alignment information from the sequences.
seqs = strrep({seed_seqs.Sequence},'.','');
names = {seed_seqs.Header};
Now align all the proteins to the HMM profile.
fprintf('Aligning sequences ') scores = zeros(numel(seqs),1); aligned_seqs = cell(numel(seqs),1); for sn=1:numel(seqs) fprintf('.') [scores(sn),aligned_seqs{sn}]=hmmprofalign(hmm_7tm,seqs{sn}); end fprintf('\n')
Aligning sequences ................................
Next, send the results to the system web browser to better explore the new multiple alignment. Columns marked with * at the bottom indicate when the model was in a "match" or "delete" state.
hmmprofmerge(aligned_seqs,names,scores)
You can also explore the alignment from the command window; the hmmprofmerge function with one output argument places the aligned sequences into a char array.
str = hmmprofmerge(aligned_seqs); str(1:10,1:80)
ans =
10×80 char array
'YILVKAIYTLGYSVS.LMSLATGSIILCLF.RKLHCTR.NYIHLNLFLSFILRAISVLVK.DDVLYSSSG-TLH......'
'FRSVKIGYTIGHSVS.LISLTTAIVILCMS.RKLHCTR.NYIHMHLFVSFILKAIAVFVK.DAVLYDVIQESDN......'
'YNTVKTGYTIGYSLS.LASLLVAMAILSLF.RKLHCTR.NYIHMHLFMSFILRATAVFIK.DMALFNSG-EIDH......'
'FGAIKTGYTIGHSLS.LISLTAAMIILCIF.RKLHCTR.NYIHMHLFMSFIMRAIAVFIK.DIVLFESG-ESDH......'
'YLSVKALYTVGYSTS.LVTLTTAMVILCRF.RKLHCTR.NFIHMNLFVSFMLRAISVFIK.DWILYAEQD-SSH......'
'FSTVKIIYTTGHSIS.IVALCVAIAILVAL.RRLHCPR.NYIHTQLFATFILKASAVFLK.DAAIFQGDS-TDH......'
'LSTLKQLYTAGYATS.LISLITAVIIFTCF.RKFHCTR.NYIHINLFVSFILRATAVFIK.DAVLFSDET-QNH......'
'FDRLGMIYTVGYSVS.LASLTVAVLILAYF.RRLHCTR.NYIHMHLFLSFMLRAVSIFVK.DAVLYSGATLDEA......'
'FERLYVMYTVGYSIS.FGSLAVAILIIGYF.RRLHCTR.NYIHMHLFVSFMLRATSIFVK.DRVVHAHIGVKEL......'
'ALNLFYLTIIGHGLS.IASLLISLGIFFYF.KSLSCQR.ITLHKNLFFSFVCNSVVTIIH.LTAVANNQALVAT......'
Looking for Similarity with Sequence Comparison
Having a profile HHM which describes this family has several advantages over plain sequence comparison. Suppose that you have a new oligonucleotide that you want to relate to the 7-transmembrane receptor family. For this example, get a protein sequence from NCBI and extract the aminoacid sequence.
mousegpcr = getgenpept('NP_783573');
Bai3 = mousegpcr.Sequence;
This sequence is also provided in the MAT-file gpcrfam.mat.
load('gpcrfam.mat','mousegpcr') Bai3 = mousegpcr.Sequence; seqdisp(Bai3,'row',70)
ans =
22×82 char array
' 1 MKAVRNLLIY IFSTYLLVMF GFNAAQDFWC STLVKGVIYG SYSVSEMFPK NFTNCTWTLE NPDPTKYSIY'
' 71 LKFSKKDLSC SNFSLLAYQF DHFSHEKIKD LLRKNHSIMQ LCSSKNAFVF LQYDKNFIQI RRVFPTDFPG'
' 141 LQKKVEEDQK SFFEFLVLNK VSPSQFGCHV LCTWLESCLK SENGRTESCG IMYTKCTCPQ HLGEWGIDDQ'
' 211 SLVLLNNVVL PLNEQTEGCL TQELQTTQVC NLTREAKRPP KEEFGMMGDH TIKSQRPRSV HEKRVPQEQA'
' 281 DAAKFMAQTG ESGVEEWSQW SACSVTCGQG SQVRTRTCVS PYGTHCSGPL RESRVCNNTA LCPVHGVWEE'
' 351 WSPWSLCSFT CGRGQRTRTR SCTPPQYGGR PCEGPETHHK PCNIALCPVD GQWQEWSSWS HCSVTCSNGT'
' 421 QQRSRQCTAA AHGGSECRGP WAESRECYNP ECTANGQWNQ WGHWSGCSKS CDGGWERRMR TCQGAAVTGQ'
' 491 QCEGTGEEVR RCSEQRCPAP YEICPEDYLI SMVWKRTPAG DLAFNQCPLN ATGTTSRRCS LSLHGVASWE'
' 561 QPSFARCISN EYRHLQHSIK EHLAKGQRML AGDGMSQVTK TLLDLTQRKN FYAGDLLVSV EILRNVTDTF'
' 631 KRASYIPASD GVQNFFQIVS NLLDEENKEK WEDAQQIYPG SIELMQVIED FIHIVGMGMM DFQNSYLMTG'
' 701 NVVASIQKLP AASVLTDINF PMKGRKGMVD WARNSEDRVV IPKSIFTPVS SKELDESSVF VLGAVLYKNL'
' 771 DLILPTLRNY TVVNSKVIVV TIRPEPKTTD SFLEIELAHL ANGTLNPYCV LWDDSKSNES LGTWSTQGCK'
' 841 TVLTDASHTK CLCDRLSTFA ILAQQPREIV MESSGTPSVT LIVGSGLSCL ALITLAVVYA ALWRYIRSER'
' 911 SIILINFCLS IISSNILILV GQTQTHNKSI CTTTTAFLHF FFLASFCWVL TEAWQSYMAV TGKIRTRLIR'
' 981 KRFLCLGWGL PALVVATSVG FTRTKGYGTD HYCWLSLEGG LLYAFVGPAA AVVLVNMVIG ILVFNKLVSR'
'1051 DGILDKKLKH RAGQMSEPHS GLTLKCAKCG VVSTTALSAT TASNAMASLW SSCVVLPLLA LTWMSAVLAM'
'1121 TDKRSILFQI LFAVFDSLQG FVIVMVHCIL RREVQDAFRC RLRNCQDPIN ADSSSSFPNG HAQIMTDFEK'
'1191 DVDIACRSVL HKDIGPCRAA TITGTLSRIS LNDDEEEKGT NPEGLSYSTL PGNVISKVII QQPTGLHMPM'
'1261 SMNELSNPCL KKENTELRRT VYLCTDDNLR GADMDIVHPQ ERMMESDYIV MPRSSVSTQP SMKEESKMNI'
'1331 GMETLPHERL LHYKVNPEFN MNPPVMDQFN MNLDQHLAPQ EHMQNLPFEP RTAVKNFMAS ELDDNVGLSR'
'1401 SETGSTISMS SLERRKSRYS DLDFEKVMHT RKRHMELFQE LNQKFQTLDR FRDIPNTSSM ENPAPNKNPW'
'1471 DTFKPPSEYQ HYTTINVLDT EAKDTLELRP AEWEKCLNLP LDVQEGDFQT EV '
First, using local alignment compare the new sequence to one of the sequences in the multiple alignment. For instance use the first sequence, in this case the human protein 'VIPR2'. The Smith-Waterman algorithm (swalign) can make use of scoring matrices. Scoring matrices can capture the probability of substitution of symbols. The sequences in this example are known to be only distantly related, so BLOSUM30 is a good choice for the scoring matrix.
VIPR2 = seqs{1};
[sc_aa_affine, alignment] = swalign(Bai3,VIPR2,'ScoringMatrix',...
'blosum30','gapopen',5,'extendgap',3,'showscore',true);
sc_aa_affine
sc_aa_affine = 69.6000

By looking at the scoring space, apparently, both sequences are related. However, this relationship could not be inferred from a dot plot.
Bai3_aligned_region = strrep(alignment(1,:),'-',''); seqdotplot(VIPR2,Bai3_aligned_region,7,2) ylabel('VIPR2'); xlabel('Bai3');
Is either of these two examples enough evidence to affirm that these sequences are related? One way to test this is to randomly create a fake sequence with the same distribution of amino acids and see how it aligns to the family. Notice that the score of the local alignment between the fake sequence and the VIPR2 protein is not significantly lower than the score of the alignment between the Bia3 and VIPR2 proteins. To ensure reproducibility of the results of this example, we reset the global random generator.
rng(0,'twister'); fakeSeq = randseq(1000,'FROMSTRUCTURE',aacount(VIPR2)); sc_fk_affine = swalign(fakeSeq,VIPR2,'ScoringMatrix','blosum30',... 'gapopen',5,'extendgap',3,'showscore',true)
sc_fk_affine = 60.4000
In contrast, when you align both sequences to the family using the trained profile HMM, the score of aligning the target sequence to the family profile is significantly larger than the score of aligning the fake sequence.
sc_aa_hmm = hmmprofalign(hmm_7tm,Bai3) sc_fk_hmm = hmmprofalign(hmm_7tm,fakeSeq)
sc_aa_hmm = 214.5286 sc_fk_hmm = -49.1624
Exploring Profile HMM Alignment Options
Similarly to the swalign alignment function, when you use profile alignments you can visualize the scoring space using the showscore option to the hmmprofalign function.
Display Bai3 aligned to the 7tm_2 family.
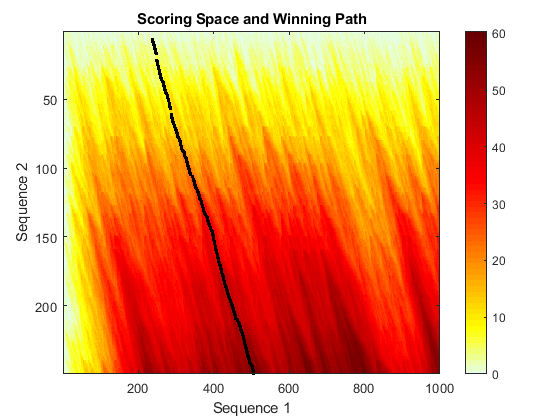
hmmprofalign(hmm_7tm,Bai3,'showscore',true); title('log-odds score for best path: Bai3');

Display the "fake" sequence aligned to the 7tm_2 family.
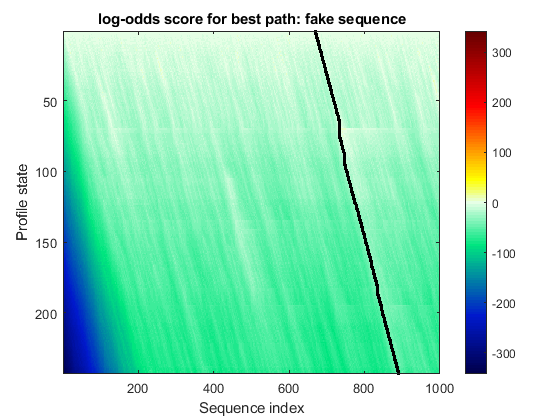
hmmprofalign(hmm_7tm,fakeSeq,'showscore',true); title('log-odds score for best path: fake sequence');
Display Bai3 globally aligned to the 7tm_2 family.
[sc_aa_hmm,align,ptrs] = hmmprofalign(hmm_7tm,Bai3); Bai3_hmmaligned_region = Bai3(min(ptrs):max(ptrs)); hmmprofalign(hmm_7tm,Bai3_hmmaligned_region,'showscore',true); title('log-odds score for best path: Bai3 aligned globally');

Align tandemly repeated domains.
naa = numel(Bai3_hmmaligned_region); repeats = randseq(1000,'FROMSTRUCTURE',aacount(Bai3)); %artificial example repeats(200+(1:naa)) = Bai3_hmmaligned_region; repeats(500+(1:naa)) = Bai3_hmmaligned_region; repeats(700+(1:naa)) = Bai3_hmmaligned_region; hmmprofalign(hmm_7tm,repeats,'showscore',true); title('log-odds score for best path: Bai3 tandem repeats');

Searching for Fragment Domains
In MATLAB®, you can search for fragment domains by manually activating the B->M and M->E transition probabilities of the HMM model.
hmm_7tm_f = hmm_7tm; hmm_7tm_f.BeginX(3:end)=.002; hmm_7tm_f.MatchX(1:end-1,4)=.002;
Create a random sequence, or fragment model, with a small insertion of the Bai3 protein:
fragment = randseq(1000,'FROMSTRUCTURE',aacount(Bai3));
fragment(501:550) = Bai3_hmmaligned_region(101:150);
Try aligning the random sequence with the inserted peptide to both models, the global and fragment model:
hmmprofalign(hmm_7tm,fragment,'showscore',true); title('log-odds score for best path: PF00002 global '); hmmprofalign(hmm_7tm_f,fragment,'showscore',true); title('log-odds score for best path: PF00002 fragment domains');


Exploring the Profile HMMs
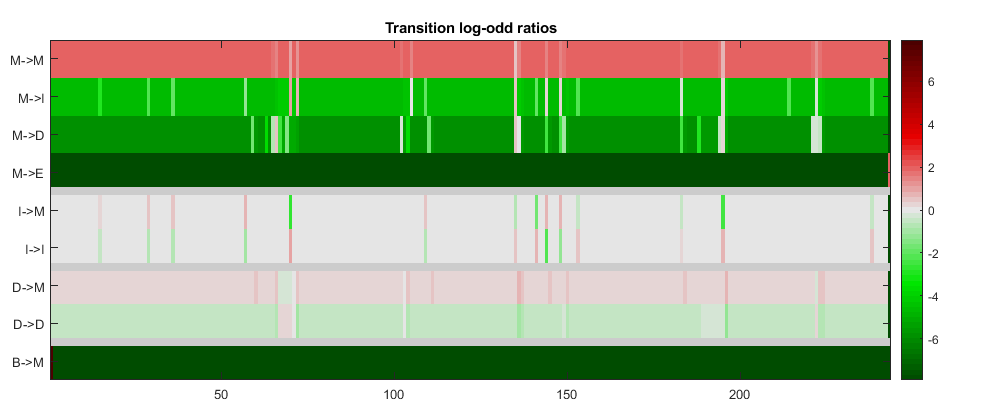
The function showhmmprof is an interactive tool to explore the profile HMM. Try right and left mouse clicks over the model figures. There are three plots for each model: (1) the symbol emission probabilities in the Match states, (2) the symbol emission probabilities in the Insert states, and (3) the Transition probabilities.
showhmmprof(hmm_7tm,'scale','logodds')


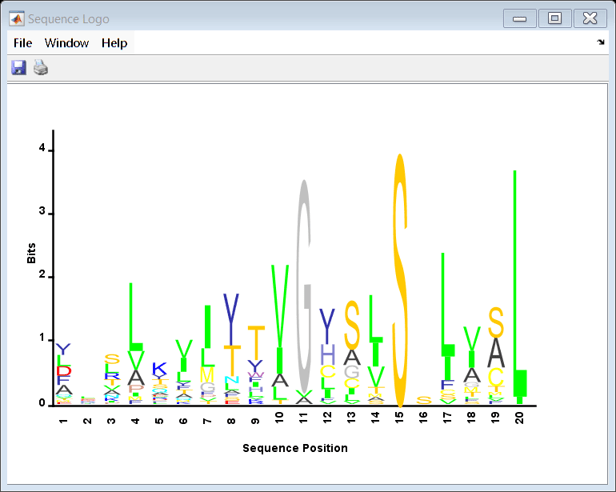
An alternative method to explore a profile HMM is by creating a sequence logo from the multiple alignment. A sequence logo displays the frequency of bases found at each position within a given region, usually for a binding site. Using the hmm_7tm sequences, consider the portion of the Parathyroid hormone-related peptide receptor (precursor) found at the n-terminus of the PTRR_Human sequence. The seqlogo allows a quick visual comparison of how well this region is conserved across the 7tm family.
seqlogo(str,'startat',1,'endat',20,'alphabet','AA')
Profile Estimation
Profile HMMs can also be estimated from a multiple alignment. As new sequences related to the family are found, it is possible to re-estimate the model parameters.
hmm_7tm_new = hmmprofestimate(hmm_7tm,str)
hmm_7tm_new =
struct with fields:
Name: '7tm_2'
PfamAccessionNumber: 'PF00002.19'
ModelDescription: '7 transmembrane receptor (Secretin family)'
ModelLength: 243
Alphabet: 'AA'
MatchEmission: [243×20 double]
InsertEmission: [243×20 double]
NullEmission: [0.0768 0.0418 0.0396 0.0305 0.0201 … ] (1×20 double)
BeginX: [244×1 double]
MatchX: [242×4 double]
InsertX: [242×2 double]
DeleteX: [242×2 double]
FlankingInsertX: [2×2 double]
LoopX: [2×2 double]
NullX: [2×1 double]
In case your sequences are not pre-aligned, you can also utilize the multialign function before estimating a new HMM profile. It is possible to refine the HMM profile by re-aligning the sequences to the model and re-estimating the model iteratively until you converge to a locally optimal model.
aligned_seqs = multialign(seqs); hmm_7tm_ma = hmmprofestimate(hmmprofstruct(270),aligned_seqs) showhmmprof(hmm_7tm_ma,'scale','logodds') close; close; % close insertion emission prob. and transition prob.
hmm_7tm_ma =
struct with fields:
ModelLength: 270
Alphabet: 'AA'
MatchEmission: [270×20 double]
InsertEmission: [270×20 double]
NullEmission: [0.0500 0.0500 0.0500 0.0500 0.0500 … ] (1×20 double)
BeginX: [271×1 double]
MatchX: [269×4 double]
InsertX: [269×2 double]
DeleteX: [269×2 double]
FlankingInsertX: [2×2 double]
LoopX: [2×2 double]
NullX: [2×1 double]

Align all sequences to the new model.
fprintf('Aligning sequences ') scores = zeros(numel(seqs),1); aligned_seqs = cell(numel(seqs),1); for sn=1:numel(seqs) fprintf('.') [scores(sn),aligned_seqs{sn}]=hmmprofalign(hmm_7tm_ma,seqs{sn}); end fprintf('\n') str = hmmprofmerge(aligned_seqs); str(1:10,1:80)
Aligning sequences ................................
ans =
10×80 char array
'YILVKAIYTLGYSVSLMSLATGSIILCLF.RKLHCTRNYIHLNLFLSFILRAISVLVKDDVLYSS---SGTLHCP-....'
'FRSVKIGYTIGHSVSLISLTTAIVILCMS.RKLHCTRNYIHMHLFVSFILKAIAVFVKDAVLYDVIQ--ESDNCS-....'
'YNTVKTGYTIGYSLSLASLLVAMAILSLF.RKLHCTRNYIHMHLFMSFILRATAVFIKDMALFNS---GEIDHCS-....'
'FGAIKTGYTIGHSLSLISLTAAMIILCIF.RKLHCTRNYIHMHLFMSFIMRAIAVFIKDIVLFES---GESDHCH-....'
'YLSVKALYTVGYSTSLVTLTTAMVILCRF.RKLHCTRNFIHMNLFVSFMLRAISVFIKDWILYAE---QDSSHCF-....'
'FSTVKIIYTTGHSISIVALCVAIAILVAL.RRLHCPRNYIHTQLFATFILKASAVFLKDAAIFQG---DSTDHCS-....'
'LSTLKQLYTAGYATSLISLITAVIIFTCF.RKFHCTRNYIHINLFVSFILRATAVFIKDAVLFSD---ETQNHCL-....'
'FDRLGMIYTVGYSVSLASLTVAVLILAYF.RRLHCTRNYIHMHLFLSFMLRAVSIFVKDAVLYSGATLDEAERLTE....'
'FERLYVMYTVGYSISFGSLAVAILIIGYF.RRLHCTRNYIHMHLFVSFMLRATSIFVKDRVVHAHIGVKELESLIM....'
'ALNLFYLTIIGHGLSIASLLISLGIFFYF.KSLSCQRITLHKNLFFSFVCNSVVTIIHLTAVANNQALVATNP---....'
Show the aligned sequences in the browser.
hmmprofmerge(aligned_seqs,names,scores)